La fibrosis quística (FQ) es una
enfermedad que afecta fundamentalmente a los pulmones y en menor medida al
hígado, páncreas e intestino. Es una enfermedad que afecta a todas las razas
pero especialmente a la raza caucásica siendo una de las enfermedades genéticas
mortales con mayor incidencia. Tiene una incidencia estimada de 1 caso cada
2500 a 3500 recién nacidos y se calcula que 1 de cada 25 individuos son
portadores sanos. El nombre de fibrosis quística hace referencia al proceso de
cicatrización (fibrosis) y formación de quistes que tienen lugar en el
páncreas. En ocasiones, esta enfermedad también recibe el nombre de mucoviscidosis.
Es una enfermedad que ataca las células epiteliales exocrinas, las personas
afectadas producen un moco espeso y viscoso, que provoca una obstrucción de los
conductos de los órganos donde se localiza. Los pacientes suelen desarrollar síntomas
clínicos en los primeros años de vida entre los que destacan: enfermedad sino-pulmonar crónica (colonización/infección
bacterianas, tos y expectoración crónica, pólipos nasales, obstrucción de vías
aéreas, etc.), alteraciones
gastro-intestinales y nutriciones (prolapso rectal, síndrome de obstrucción
intestinal distal, insuficiencia pancreática, enfermedad hepática crónica,
cirrosis biliar focal o multilobar, malnutrición, etc.) y alteraciones
asociadas a déficit de sal
(alcalosis metabólica crónica y pérdida aguda de sal). Una de las principales
características es la alta concentración de sal en el sudor de los pacientes lo
que permite un diagnóstico rápido. Actualmente, no existe ningún tratamiento
curativo de la FQ aunque existen tratamientos paliativos de los síntomas
clínicos.

La FQ está cuasada
por mutaciones en el gen CFTR (del
inglés cystic fibrosis transmembrane conductance regulator). Este gen está
situado en el cromosoma 7 y fue el secuanciado por primera vez en 1989 siendo
el primer gen humano identificado sin que se conociese en el momento la
proteína que codificaba. La proteína fue posteriormente identificada y se
descubrió que era un transportador iónico de cloro regulando su transporte a
través de las membranas apical de células epiteliales. Es un gen de gran tamaño
y en la actualidad se han descrito más de 1800 mutaciones en todo el gen
capaces de causar la aparición de la FQ con frecuencias individuales variables
siendo la mayoría de estas mutaciones pequeñas deleciones. El tipo de herencia
de la FQ es autosómico recesivo por
lo que los 2 alelos deben estar mutados para que se desarrolle la enfermedad.
Debido al gran número de mutaciones causantes de FQ el diagnóstico molecular es
complicado ya que además hay que tener en cuenta que la distribución de los
alelos varía entre las distintas poblaciones por lo que los test genéticos
deben tener este hecho presente. La penetrancia de la enfermedad es variable
dependiendo de los alelos presentes en cada paciente, y a su vez, la expresión
de cada alelo depende del entorno y del resto del genoma de cada paciente.
Actualmente a los niños nada más nacer
se les hace un diagnóstico genético mediante secuenciación del gen CFTR para
saber si tienen la enfermedad, ya que es una enfermedad tratable. Cuando antes
comienza el tratamiento, mayor calidad de vida y mayor longevidad.
· Diagnóstico molecular indirecto:
básicamente realizado a través de análisis de ligamiento.
Se lleva a cabo mediante:
· Diagnóstico molecular directo: podríamos
secuenciar el gen CFTR, pero lo que está a la orden del día es la detección de
las mutaciones, que básicamente se realiza a partir de dos estrategias:
· Rastreo de mutaciones: técnicas de detección de
mutaciones, pero sin identificación de la mutación. Algunas de las más usadas
en este proceso son la electroforesis
en gel con gradiente de desnaturalización, así como algunas
variantes de PCR. Un ejemplo de esto es
la detección de la mutación ΔF508. Esta detección se realiza gracias a una
enzima de restricción que corta al alelo sano que no presenta la detección
mencionada, lo que permite que el producto de PCR tras la amplificación sea
menor en el alelo sano que en el que presenta la mutación, viéndose así la
diferencia.
· Identificación de mutaciones: técnicas basadas en
hibridación específica con el alelo mutado. Por ejemplo, los kits ASO:dot blot,
OLA (ensayo de ligación de
oligonucleótidos), o el ensayo más tradicional de southern blot.

Las parejas que están atravesando un
embarazo o tienen planes respecto de la gestación, pueden ser evaluadas en
busca de mutaciones del gen CFTR, con el objeto de determinar las
probabilidades de que su hijo nazca con fibrosis quística. La prueba se suele realizar
en uno de los padres o en ambos y, en caso de detectarse un riesgo elevado de
FQ, se efectúa también en el feto. Debido a que el diagnóstico prenatal no habilita formas
de tratamiento superiores o alternativas, la principal razón por la que se
lleva a cabo es, en la práctica, proporcionar la posibilidad del aborto en
caso de que el feto presente la enfermedad. La prueba para fibrosis quística en
parejas se ofrece de manera generalizada en países como los Estados Unidos y
el Colegio Americano de Obstetras y Ginecólogos (ACOG, por sus siglas en
inglés) recomienda la prueba en parejas que poseen un historial de FQ entre sus
familiares directos o parientes cercanos, así como también en aquellas con
riesgo elevado debido a su filiación étnica. Como
consecuencia de que no todas las mutaciones conocidas son detectadas por las
pruebas corrientes, un resultado negativo no garantiza que el niño vaya a estar
libre de la enfermedad. Por
otro lado, dado que las mutaciones sondeadas son necesariamente aquellas más
comunes en los grupos de más alto riesgo, las pruebas en etnias de bajo riesgo
son menos exitosas, ya que las mutaciones más extendidas en estos grupos son
menos frecuentes en la población general.
Un número variable de
mutaciones son relativamente comunes y se han encontrado con frecuencias
variables en distintos grupos étnicos. Para la mayoría de estas mutaciones “comunes”,
hay suficientes pacientes como para realizar análisis de sus consecuencias clínicas
y su efecto patogénico sobre la función de la proteína se ha estudiado in
vitro. Los efectos de las mutaciones comunes sobre la proteína en la célula
se han clasificado en 5 grandes grupos:
Clase
1,
con ausencia de producción de la proteína (ejemplo, p.Gli542X o p.G542X)
Clase
2,
con defecto de procesamiento de la proteína (ejemplo, p.F508del)
Clase
3,
con defecto en la regulación del canal (ejemplo, p.Gli551Asp o p.G551D)
Clase
4,
con defecto en la conductancia (ejemplo, p. Arg117His o p.R117H)
Clase
5,
por alteración del splicing o “empalme” (ejemplo c. 621+1G>T).
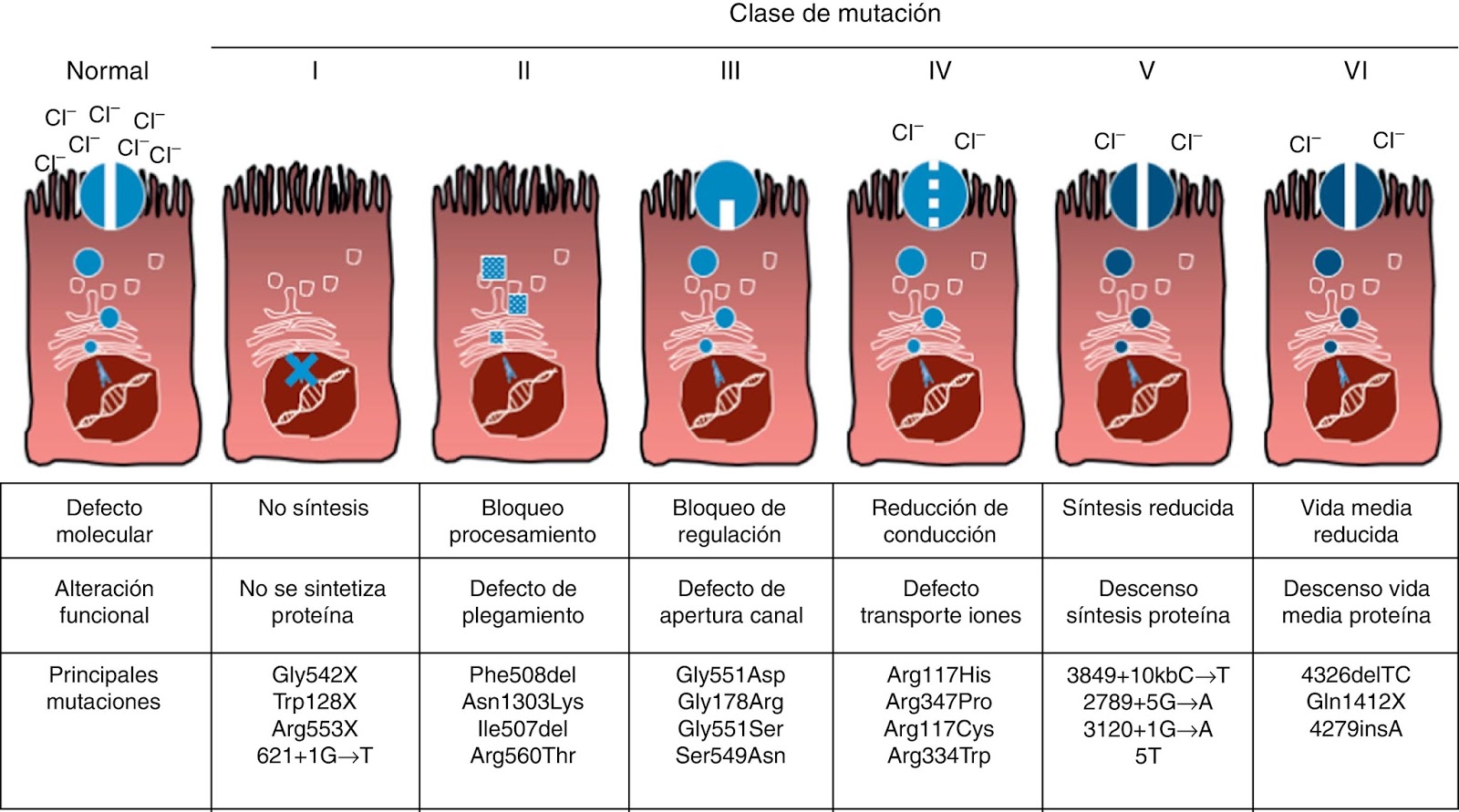
En las mutaciones
clases 1 y 2, hay ausencia de la proteína en la membrana celular. En contraste,
en las mutaciones clases 3 y 4 pueden resultar en la presencia de una proteína
con cierta actividad residual, pero el defecto funcional suele ser más marcado
en las mutaciones clase 3. Las mutaciones clase 5 tienen efecto variable, según
el tipo de mutación. Las mutaciones clases 1, 2 y 3, se consideran “severas”,
las clase 4, “leves” y las clase 5, como se señaló, tienen consecuencias
variables. El resto de las mutaciones (más de 1600) han sido descritas en uno o
muy pocos pacientes, lo que limita la posibilidad de correlacionarlas con las consecuencias
clínicas. Para la mayoría de ellas, sus efectos deletéreos no han sido
claramente demostrados in vitro. Así como se han descrito muchas mutaciones
patogénicas, también hay centenas de polimorfismos o variantes normales. Con el
uso cada vez más frecuente de screening preconcepcional o neonatal para diagnóstico
presintomático, paneles diagnósticos que incluyen gran número de mutaciones,
mayor acceso a secuenciación directa y otros métodos de búsqueda de rearreglos genómicos
(inserciones, deleciones, inversiones, etc), se ha hecho cada vez más
imperativo el contar con información que permita claramente distinguir entre
variantes normales y mutaciones causantes de enfermedad. En este sentido, el consorcio
internacional de análisis genético de FQ (Cystic
Fibrosis Genetic Analysis Consortium) ha iniciado un estudio denominado Clinical and Functional TRanslation of CFTR
o CFTR2, destinado a correlacionar todas las mutaciones con información
completa, actualizada y revisada por expertos sobre los aspectos funcionales y
las consecuencias clínicas de toda mutación listada en su base de datos (http://www.genet.sickkids.on.ca/cftr/app).
Esto, sin duda, será de gran utilidad para los clínicos.
Para
aclarar cualquier duda, si quiere más información o si quiere solicitar una
consulta, no dude en contactar con las consultas externas del Hospital Dr.
Gálvez en www.hospitalgalvez.com, por correo electrónico en la dirección consultas@hospitalgalvez.com o llamando al teléfono 952062808.

No hay comentarios:
Publicar un comentario