La
enfermedad de Charcot-Marie-Tooth (CMT) es una neuropatía hereditaria
sensitivo-motora (NHSM), descrita de forma independiente por Charcot y Marie en
París y por Tooth en Londres en 1866; se transmite en forma autosómica
dominante (AD) en
la mayoría de los casos, pero también en forma autosómica recesiva (AR) y
ligada al cromosoma X. Presenta una incidencia estimada de 1 por cada 2,500 nacidos
vivos y una prevalencia de 17 a 25 por 100,000 habitantes, por lo que es considerado
el desorden neuromuscular hereditario más común. Presenta penetrancia incompleta,
heterogeneidad genética y expresividad variable. Es causada por mutaciones
específicas en uno o varios genes implicados en la estructura, formación y
mantenimiento de la vaina de mielina o de los axones neuronales. La enfermedad
CMT es un desórden genéticamente heterogéneo con un fenotipo clínico común. Las
principales características de CMT son debidas a alteración en la neurona
motora inferior acompañada de signos sensoriales, que en combinación ocasionan
una neuropatía neurosensorial. En el fenotipo clásico, los síntomas inician en
la primera o segunda década de la vida, con un curso lento y progresivo. Sin
embargo, la edad de inicio, progresión y severidad varían dependiendo de la forma
de CMT, el gen afectado, así como el tipo de mutación. Los primeros signos son:
debilidad y atrofia muscular con hiporreflexia o arreflexia en extremidades inferiores.
Posteriormente, debido a la cronicidad de la neuropatía motora presentan: pie
cavo, dedos en martillo, dificultad para caminar y correr, así como marcha equina
o steppage. También pueden presentar disminución de la percepción del dolor,
temperatura o propiocepción en miembros inferiores, mano en garra, temblor en
manos, calambres, pies fríos, hiperqueratosis plantar y acrocianosis. Sin
embargo, ciertas características clínicas son difícilmente distinguibles entre
formas axonal y desmielinizante. Los cambios electrofisiológicos son
detectables desde la infancia, mostrando una prolongación de la latencia motora
distal en los primeros meses de vida y más tarde una alteración en la velocidad de conducción nerviosa (VCN).

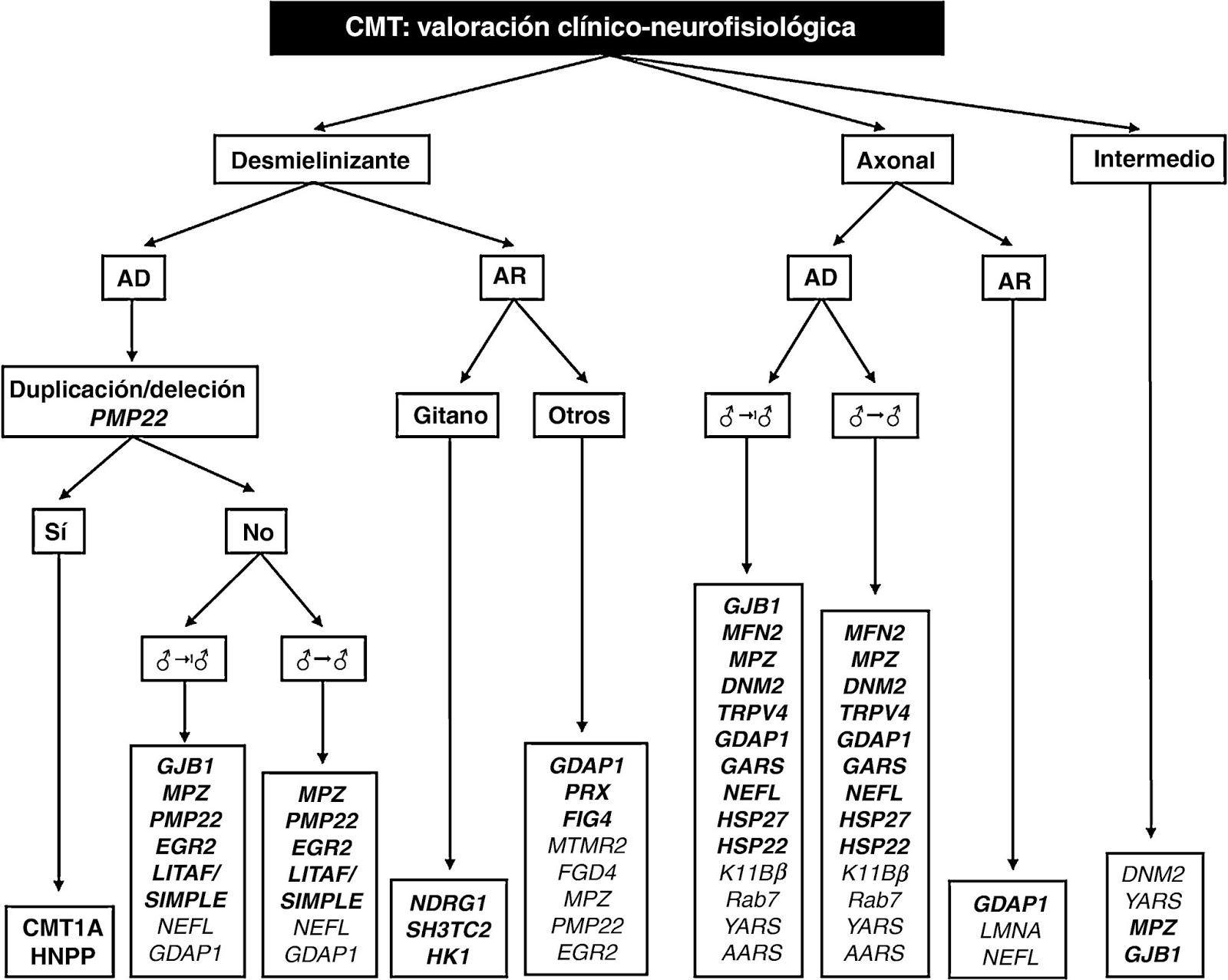
Clasificación
Inicialmente
se desarrolló una clasificación de las CMT en base al fenotipo clínico, electrofisiológico
y patrones de herencia. Después, con la identificación de los genes
involucrados, se ha mejorado dicha clasificación, ya que la subdivisión de los tipos
de CMT se realiza en base a los genes causantes, complementando así la
descripción clínica y electrofisiológica básica. De acuerdo a la velocidad de
conducción nerviosa (VCN) motora y a las características de la biopsia de
nervio, la enfermedad se puede dividir en dos grandes grupos: Forma desmielinizante: caracterizada
por una VCN motora disminuida; por lo general, de 5 a 30 m/s y mielina anormal
en biopsia de nervio (CMT1 si es AD, CMT4 si es AR). Forma axonal: donde la VCN motora es conservada o ligeramente
disminuida, con potenciales de amplitud reducida, así como signos de
degeneración y regeneración axonal crónica en biopsia de nervio (CMT2).
Tipos más comunes de CMT (ver tabla para más información):
CMT1: es una neuropatía periférica
desmielinizante con herencia AD18, caracterizada por debilidad y atrofia
muscular distal, disminución de la VCN motora (5 a 30 m/s), pie cavo, dedos en
martillo e hipoacusia. El 5% de los afectados llega a ser dependiente de silla
de ruedas. Existen 6 subtipos de CMT1, los cuales son difíciles de distinguir
clínicamente, pero identificables a través del estudio molecular. La CMT1A es
el tipo más común de CMT con una prevalencia de 1: 5,000 habitantes, representa
del 40 al 50% de todos los casos y del 70 al 80% dentro del subtipo CMT1. Esta
patología es causada por mutaciones en el gen PMP22.
CMT2: es una neuropatía periférica
axonal no desmielinizante con herencia AD, caracterizada por debilidad y
atrofia muscular distal. Por lo general la VCN motora es normal o ligeramente
disminuida (35 a 48 m/s). El fenotipo clínico es similar a CMT1; sin embargo, el
curso de la enfermedad tiende a ser más benigno, con menor afectación sensorial.
Existen al menos 5 subtipos de CMT2 de acuerdo a los hallazgos moleculares, los
cuales son difíciles de distinguir por características clínicas. Los genes con más
frecuencia asociados a esta patología son MFN2, Cx32 y MPZ5.
CMT3: se le conoce como neuropatía Dejerine-Sottas (DJS), la
cual fue originalmente descrita como una neuropatía desmielinizante de herencia
AR o AD, inicio en la infancia, afectación grave, retardo psicomotor, VCN
motora muy disminuida (< 10 m/s), aumento de proteínas en líquido cefalorraquídeo
(LCR), hipertrofia de nervio, desmielinización y presencia de bulbos de cebolla
en biopsia de nervio. En la actualidad se considera la neuropatía DJS como la
forma más grave del tipo desmielinizante. Esta entidad es debida a mutaciones en
los genes PMP22, MPZ y EGR2.
DI-CMT: la forma intermedia con
patrón de herencia AD de CMT (DI-CMT) se caracteriza por el fenotipo clásico de
CMT más ambos hallazgos anatomopatógicos en la biopsia de nervio: mielina
anormal y axonopatía. La VCN motora es de 25 a 50m/s, por lo que se considera intermedia
entre la observada en CMT1 y CMT2. Los genes que se han asociado a esta forma
de CMT son MPZ y GJB1.
CMT4: es una neuropatía
neurosensorial axonal y desmielinizante progresiva que muesta un patrón de herencia
AR con una VCN motora de 20 a 30m/s y manifestaciones de inicio temprano como
retardo motor, debilidad y atrofia de músculos distales con posterior propagación
a músculos proximales. Las mutaciones en el gen FIG4 son responsables de
algunas variantes de CMT4.
CMTX1: es el segundo tipo más
frecuente de CMT (10%), con herencia ligada al cromosoma X, se caracteriza por
ser una neuropatía neurosensorial de moderada
a grave
en hombres; las mujeres portadoras son; por lo general, asintomáticas o
muestran un fenotipo leve, aunque suelen presentar hipoacusia neurosensorial.
Se
reconocen
5 subtipos de CMT ligados al cromosoma X. Los casos recesivos de CMTX se
presentan con sordera, retraso mental y encefalomielitis. Esta entidad es
causada por mutaciones en el gen GJB1.
En la
Tabla 1 se propone una clasificación más completa y detallada de la enfermedad
de CMT con base a criterios clínicos, electrofisiológicos y moleculares.

Bases moleculares
Hasta
la fecha, por medio de la técnica de ligamiento genético se han identificado más
de 30 genes asociados a esta enfermedad, expresados tanto en las células de Schwann
como en los axones neuronales, que causan fenotipos solapados, entre ellos los principales son: PMP22, MPZ, NEFL, GJB1, MFN2 y EGR2.
PMP22: se localiza en el cromosoma
17p11.2-p12 y codifica para la proteína 22 de mielina periférica (PMP22), una glicoproteína
que consta de 160 aminoácidos, con peso molecular de 22 kDa y cuatro dominios
transmembrana glucosilados en el extremo amino terminal, cuya función es
regular la síntesis de mielina, proporcionando principalmente estabilidad y
grosor a la vaina de mielina. Una duplicación en tandem de 1.5 Mb en dicho gen
está asociada a CMT1-A.
MPZ: se localiza en el cromosoma
1p36, codifica para la proteína cero de mielina (MPZ) la cual es una glicoproteína
transmembrana de 29 kDa; es considerada la mayor proteína estructural de
mielina del SNP. Está conformada por un dominio extracelular, un dominio transmembrana
y un dominio citoplasmático; el dominio extracelular es importante para la
formación de capas compactas de mielina de células de Schwann. Las mutaciones
en el gen MPZ se asocian con CMT1-B.
NEFL: se localiza en el cromosoma
8p21 y codifica para la proteína de neurofilamentos de cadena ligera (NFL), la
cual tiene un peso de 62 kDa. Participa de forma importante en el ensamblaje y
mantenimiento de neurofilamentos, así como en el control del crecimiento y calibre
de los axones mielinizados. Las mutaciones en este gen son responsables del
subtipo CMT2E y CMT1F.
GJB1: localizado en el cromosoma
Xq13.1, codifica para la proteína beta 1 de uniones gap llamada conexina32
(Cx32), la cual es expresada en oligodendrocitos, SNC y en células de Schwann.
Las uniones gap, permiten la difusión de pequeñas moléculas a través de las
vainas de mielina, por lo que se propone que mutaciones en este gen impiden la
comunicación celular normal, dando como resultado disfunción en la mielinización
de las células de Schwann. Mutaciones en este gen han sido asociadas a CMTX1.
MFN2: está localizado en el
cromosoma 1p35-p36. Codifica para la proteína mitofusina 2 (MFN2). Las mitofusinas
son GTPasas de membrana mitocondrial, constituidas por un dominio N-terminal y
un domino C-terminal corto expuesto al citosol. Mutaciones en este gen se
asocian a CMT2A.
EGR2: localizado en 10q22-q23,
codifica para un factor de transcripción llamado proteína 2 de respuesta temprana
al crecimiento, con función en la mielinización de nervios periféricos, específicamente
en células de Schwann y cuyas mutaciones son responsables del tipo D-CMT.
Conclusiones y Perspectivas
La
degeneración mielínica desempeña un papel importante en la aparición de la
enfermedad en sus distintas formas clínicas. La regulación del proceso de mielinización-desmielinización
es compleja, y mutaciones en genes involucrados pueden causar efectos distintos
en la célula de Schwann en su interacción con el axón, lo que provoca
alteraciones significativas en la fisiología nerviosa. La clasificación de la
enfermedad de CMT es extensa, compleja y constantemente sometida a cambios debido
al descubrimiento de nuevas mutaciones en genes asociados. La historia natural
de la enfermedad sigue un curso lento y progresivo con heterogeneidad tanto
clínica como genética, por lo cual los datos clínicos y electrofisiológicos son
indispensables para la orientación hacia un diagnóstico oportuno. El poder
identificar el subtipo de CMT en los pacientes podría a futuro permitir el
desarrollo de estrategias más selectivas en estas variantes de la enfermedad,
como la nanotecnología o terapia génica y de esta manera ofrecer un tratamiento
personalizado.
Para aclarar cualquier duda, si quiere más información o si quiere
solicitar una consulta, no dude en contactar con las consultas externas del Hospital
Dr. Gálvez en www.hospitalgalvez.com, por correo electrónico en la
dirección consultas@hospitalgalvez.com o llamando al teléfono 952062808.

No hay comentarios:
Publicar un comentario